RStudio容器使用及MSI数据处理流程
Version:2.1
- 2025/11/19
- 更新RStudio标准连接
- 更新不同版本R创建标准容器流程
- 更新过时的Cardinal包函数
一、MSI数据
常用的数据类型有哪些?
在质谱成像领域内,你大概率会见到的两种文件格式:
● Raw文件格式
● imzML原始数据文件格式
事实上不同厂家的质谱仪器生产出来的Raw质谱数据是不一样的,主流公司的数据格式如下表所示。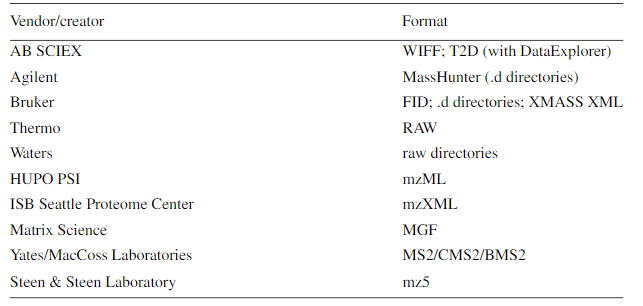
如何从Raw数据转换成imzML数据?
理论上我们只需要知道扫描斑点数信息,就可以直接从Raw文件中计算出质谱成像所需要的空间坐标信息。 本文侧重点在于对imzML文件的预处理,对于Raw文件可以用以下的链接提供的软件来进行Raw to imzML的转换,试着使用他给出的示例文件来进行练习。
https://www.ms-imaging.org/imzml/software-tools/raw-to-imzml-converter/
二、RStudio标准连接
RStudio连接办法
R部署在服务器8787端口,登录账号密码和远程桌面密码一致(未更改的话应该是姓名,姓名+123)。如果还未更改第一次登陆后就需要自己修改为更复杂的密码。
进入后可以直接使用,其中文件的使用在下一部分阐述,使用后在右上角登出。
注意:8787中R已经预装了cardinal,只需要library(cardinal)就可以正常使用了。
登录后默认处于主机中登录用户个人的home文件夹,有你存放在Share_Space和R_Share等的文件。
使用不同版本的R语言
注意:本部分仅提供给要使用特定版本的R语言的同学,不需要可以跳过。
这里可以看到已经存在一个R_EXAMPLE标准容器:
要创建自己版本的容器,从标准容器中继续点Duplicate复制容器:
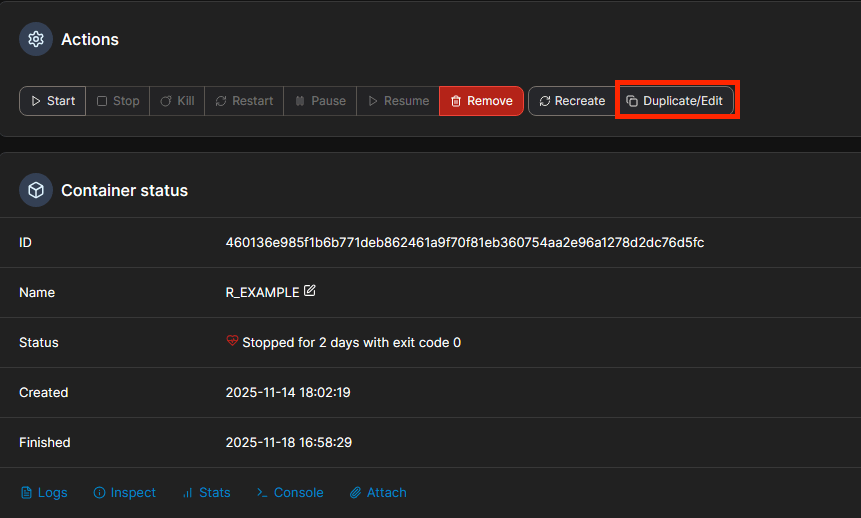
这里要改动的有如下几点,其中文件夹位置具体看下方R的文件共享部分:
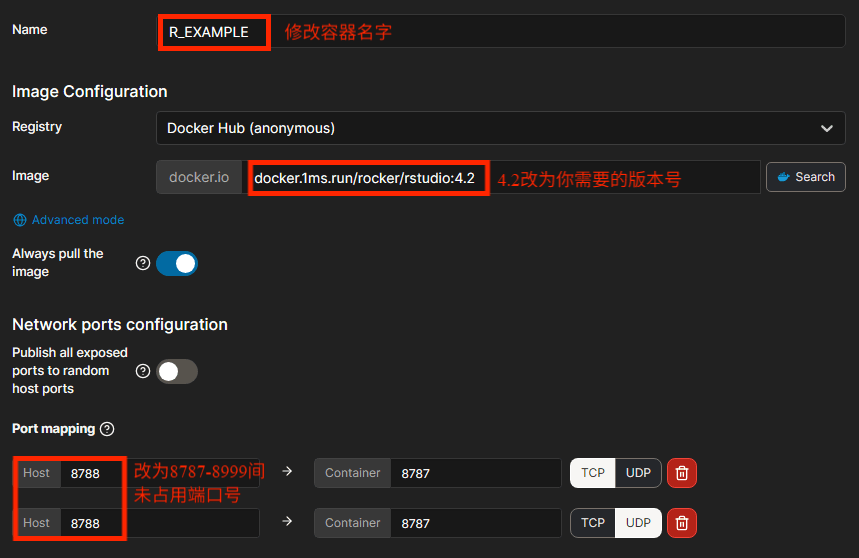
以及:
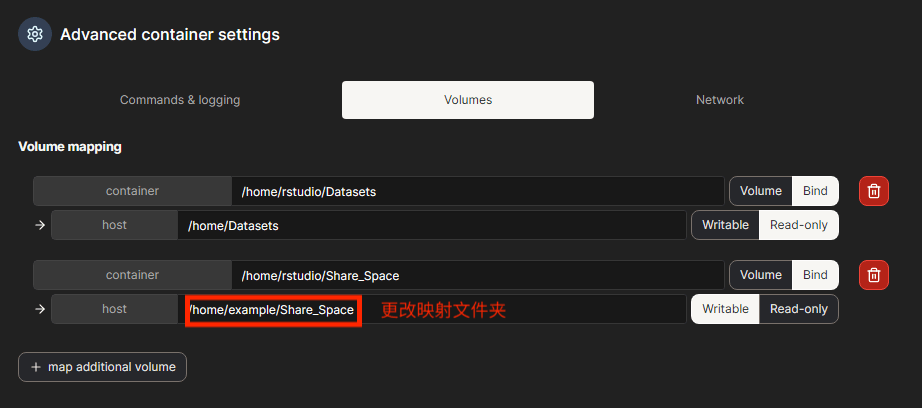
进入R语言对应的版本的端口即可,注意下图的8789应为你所修改后的端口号:
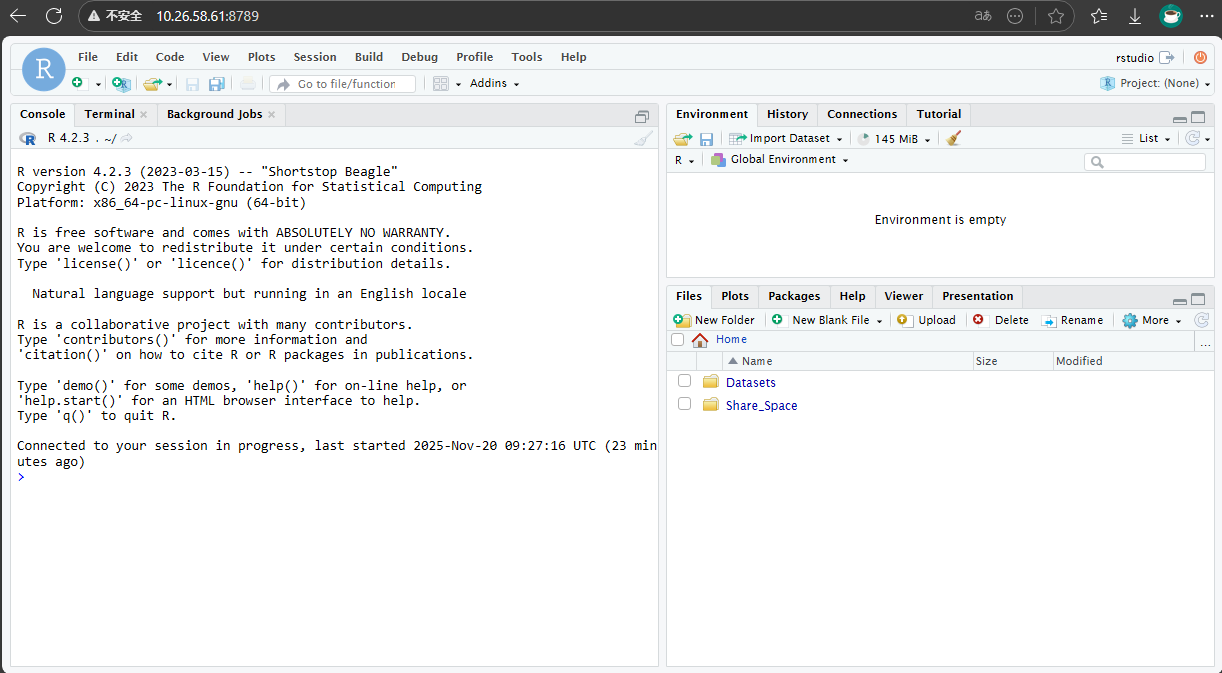
R容器的文件共享
R的工作目录默认下应该能看到如下内容:
host文件夹连接了主机的你个人的home文件夹。
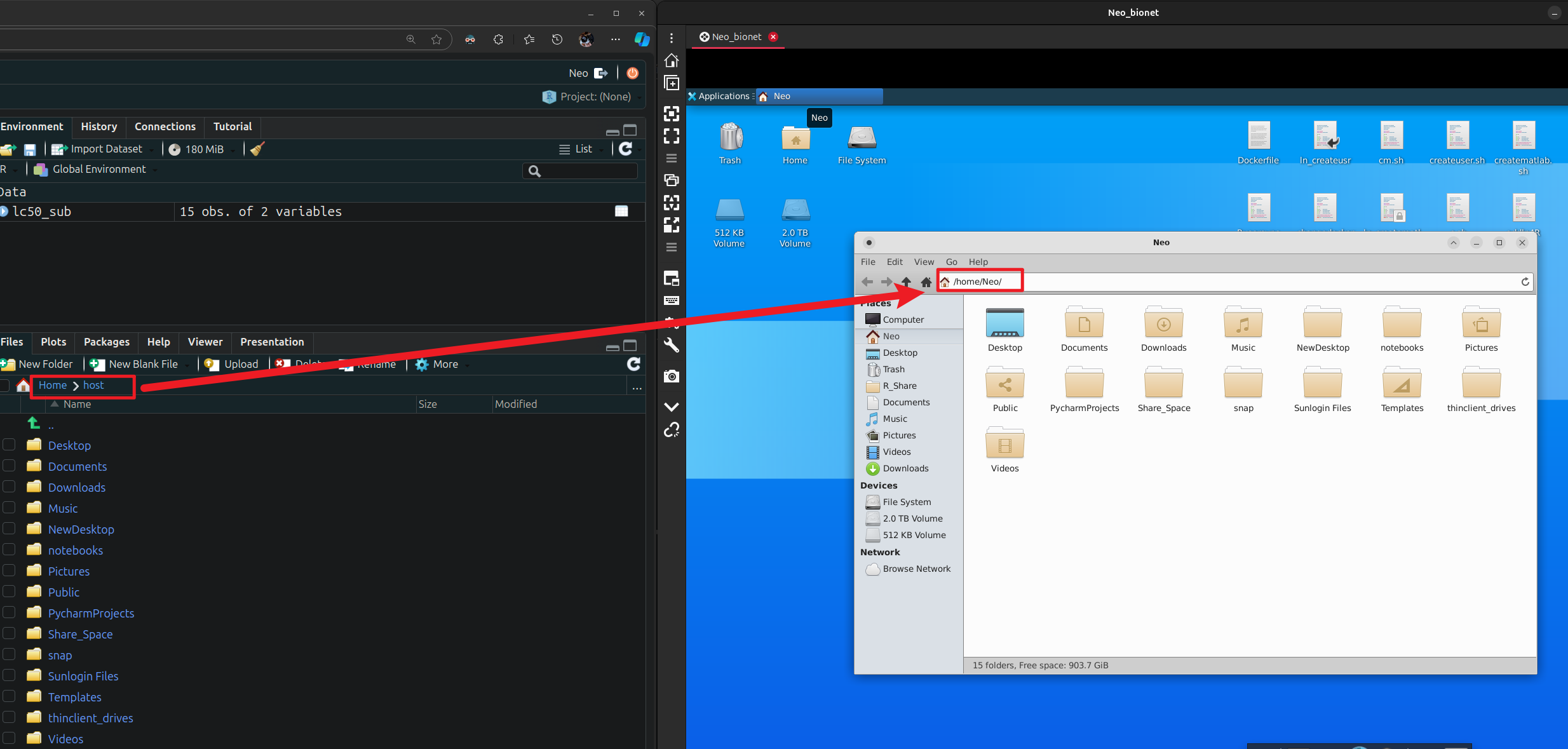
假设你主机上在你的
1 | |
文件夹下有文件A,你在R studio中调用的时候就是:
1 | |
简单来说就是将主机下:
1 | |
映射到了
1 | |
所有在主机上路径为/home/用户名/应替换为/home/Neo/host/。
Datasets文件夹连接了NAS的数据中心,与主机上/home/Datasets文件夹是同一个文件夹。
假设你在主机上NAS数据中心路径是这样的:
1 | |
那么在R studio中,应该是:
1 | |
简单来讲明就是我们将主机上:
1 | |
映射到了:
1 | |
所有路径/home/Datasets/应替换为/home/个人用户名/Datasets`。
我们举个例子:
假设你上传了数据到了此路经下:
想要在R中引用,那么我们可以在R中看到这样的:
那么在R_Studio中,代码调用的路径应该是:
1 | |
三、imzML数据预处理(Cardinal)
Cardinal 原生支持读写 imzML(包括 “continuous” 和 “processed” 类型),当然我们要处理的都是每个频谱具有不同的m/z值的 “continuous” 类型数据。
Cardinal 3.6引入了一套简单的新数据结构如下,用于组织MS成像实验的数据。
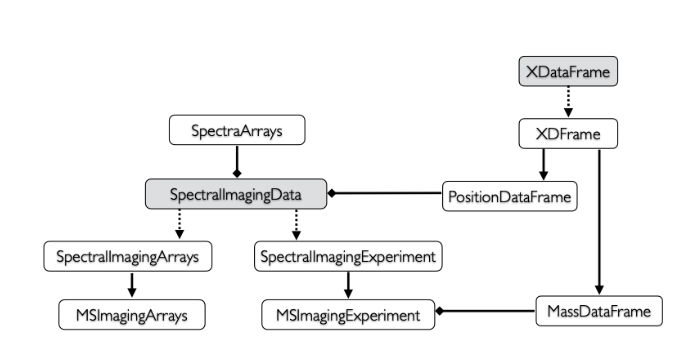
MSImagingArrays:Continuous (连续) 模式读取后格式,以下简称msa。数据存储: 在连续模式下,每个像素点的质谱数据(m/z值和对应的强度值)都完整地、独立地存储。这意味着每个像素的光谱都是一个完整的数据列表。
MSImagingExperiment:Processed (处理过) 模式读取后格式以下简称mse。数据存储: 在处理模式下,数据经过了“binning”处理。首先,会创建一个统一的、全局的m/z轴(一个m/z值的列表)。然后,每个像素的质谱数据不再存储具体的m/z值,而是只存储与全局m/z轴对应的强度值。如果某个像素在某个m/z值上没有信号,强度值就记为零。 数据变得更加规整,像一个大的数据矩阵,其中行代表像素,列代表统一的m/z通道。
理论上caldinal包提供了强制转换函数as(),但是会损失比较多的信息,所以需要预处理实现转换。
Cardinal包安装
imzML文件的数据处理有很多不同的方式,在熟悉流程之后,你可以自己编写脚本来实现。
本文使用Cardinal包来进行预处理。Cardinal包支持MALDI和DESI的MSI工作,可以对生物样品进行基于质谱实验的统计分析。
在开始前,你需要自己安装好RStudio或其他IDE。安装Cardinal包,使用以下命令:
1 | |
别忘了把刚安装好的包加载上:
1 | |
到这里如果没有报错,说明安装成功了,你可以查看官方文档来进行进一步的了解。
1 | |
读取imzML文件
1 | |
通过spectraData函数你可以初步了解你的数据,一般情况下一个对象必须至少有两个为“mz”和“intensity”的数组,分别是m/z数组和强度数组的列表。
1 | |
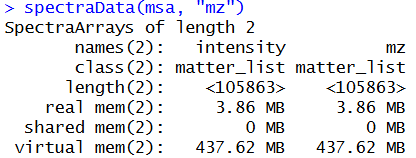
我们可以读取一些数据出来查看:
1 | |
你也可以查看实验元数据:
1 | |
运行后会给你提供三列,包含像素的x轴、y轴以及批次信息。
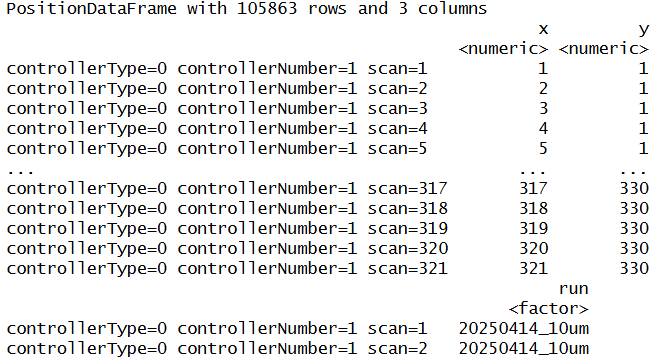
上面展示的也许不太直观,你可以通过plot来可视化查看质谱:
1 | |
其中i后面跟的是像素,这里展示了第一个像素和第五个像素的图片,用superpose=TRUE的参数重合对比。值得一提的是,根据刚才查到的元数据,图像是321x330大小,也就是说此时我的第322个像素等效于以下这种用像素坐标来指定的方式:
1 | |
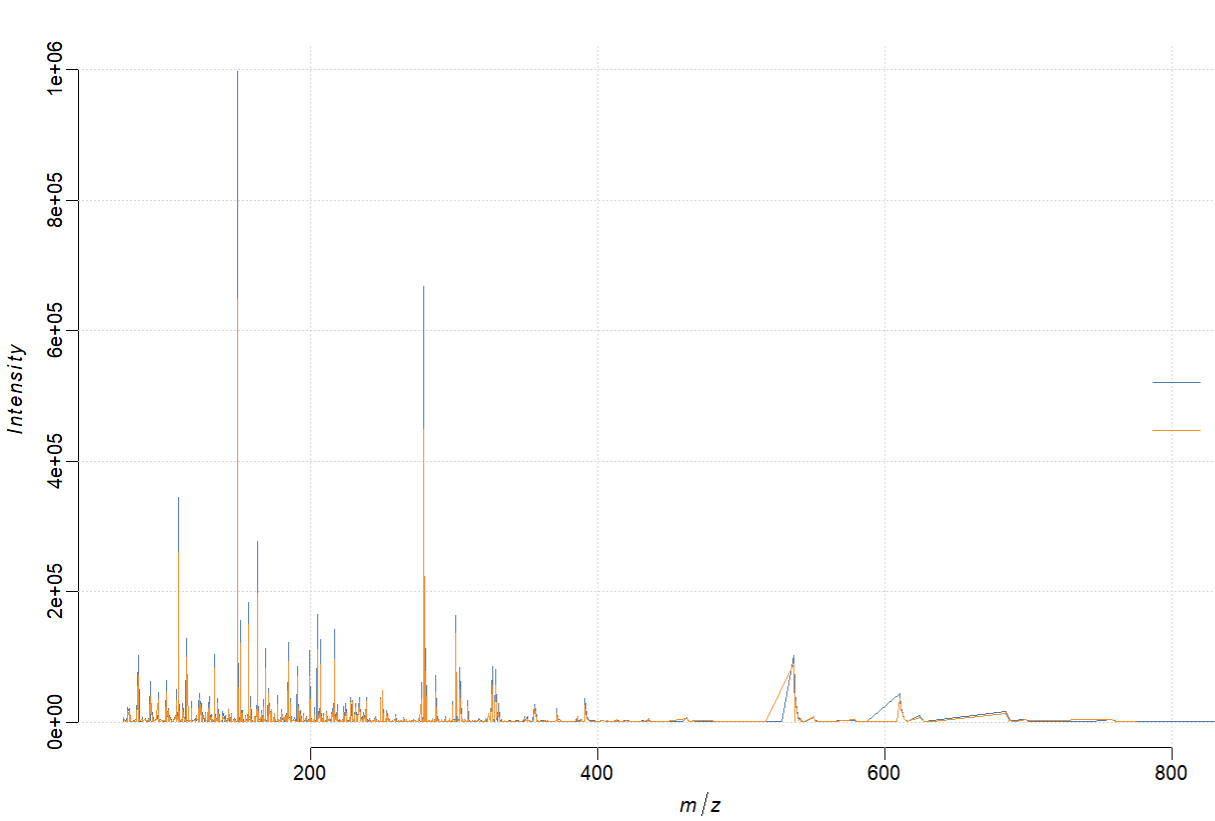
大致了解你手中的imzML数据后,可以开始进行数据预处理了。
预处理流程
归一化处理
支持的归一化方法包括:
method="tic"执行全离子电流(TIC)归一化method="rms"执行均方根(RMS)归一化method="reference"将光谱归一化为参考特征
1 | |
TIC归一化是最常见的方法。
高斯平滑处理
手册里提供了不同的平滑处理方式,使用gaussion就可以。
1 | |
基线校准
校准基线到强度为0附近。
1 | |
对齐校准
剖面光谱的对齐可以用来初步校准。
1 | |
这里process包含了(1)峰检测,(2)所有光谱上的峰值对齐,(3)滤波峰,实际使用中如果不需要其中的步骤可以根据手册只使用其中部分步骤。
峰合并分组
合并相近的峰以减少冗余。
1 | |
改变resolution可改变合并的精度,但可能丢失一些峰。需要你根据实际情况调整。
全流程处理
实际使用时cardinal包提供了队列化处理,你可以一次性提交整个流程:
1 | |
查看处理结果
由于处理后已经由msa转变成mse,你可以使用image函数来比较预处理前后的图像结果了。
输出
注意修改mse_queue部分即可
1 | |