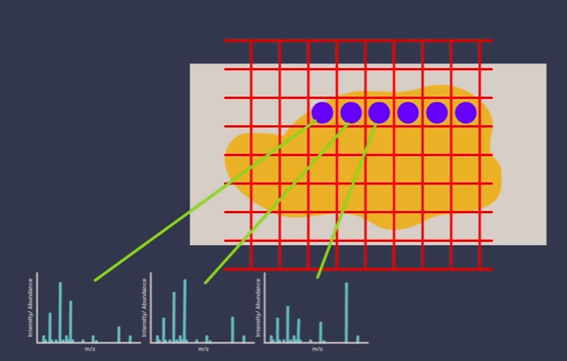
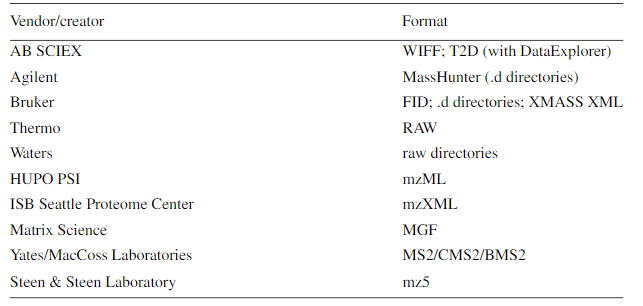
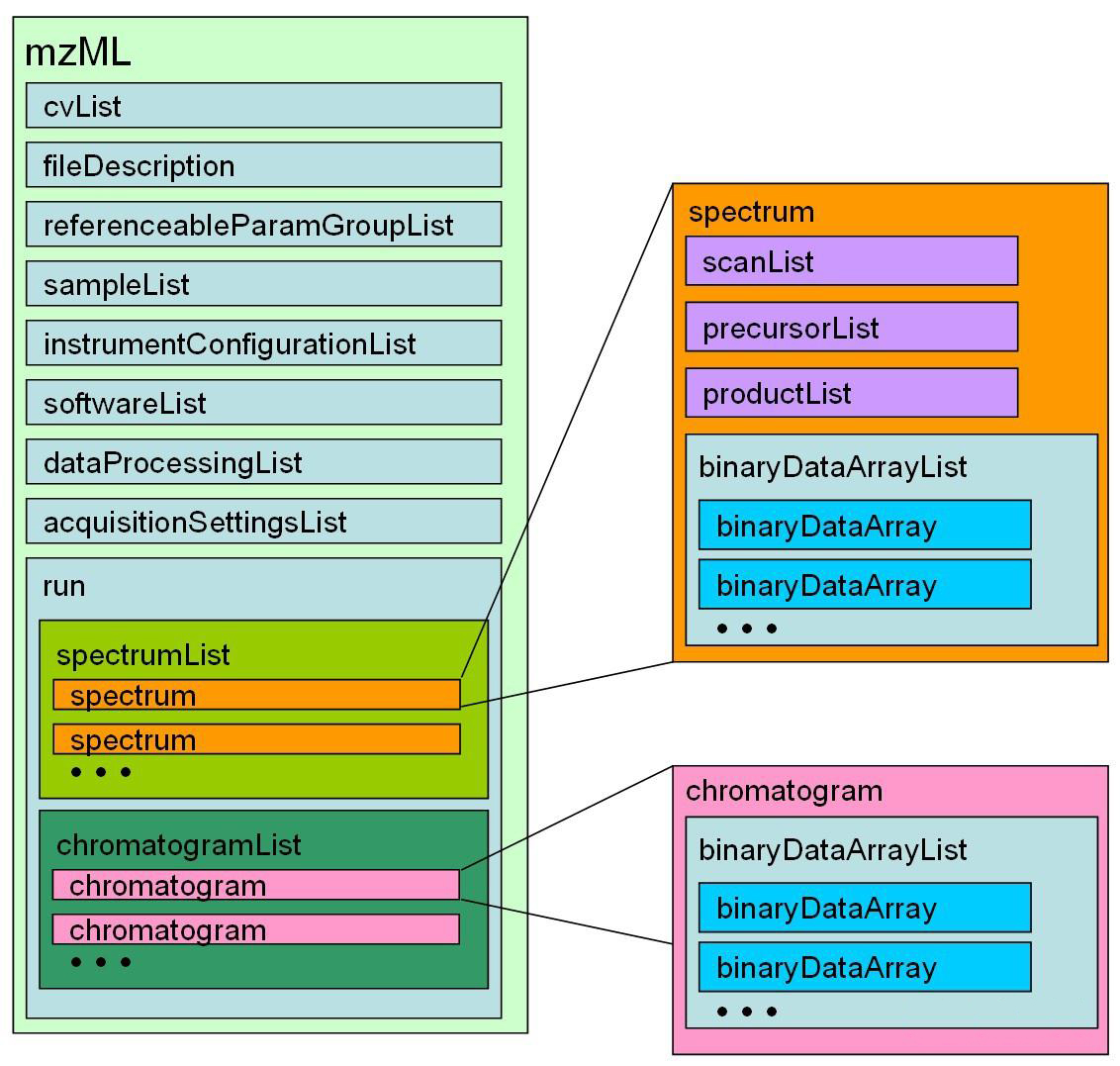
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
| import os
import numpy as np
import pandas as pd
from scipy import sparse
from scipy.ndimage import gaussian_filter1d
from pyimzml.ImzMLParser import ImzMLParser
from pyimzml.ImzMLWriter import ImzMLWriter
import csv
input_path = '/Users/ForRiver/OneDrive/Desktop/Pre/BIONET/preprocess/slice_1.imzML'
output_path = '/Users/ForRiver/OneDrive/Desktop/Pre/BIONET/preprocess/compare-neg/gz4_5.imzML'
output_dir = "/Users/ForRiver/OneDrive/Desktop/Pre/BIONET/preprocess/compare-neg/export-gz4_5/"
sorted_peaklist_file = '/Users/ForRiver/OneDrive/Desktop/Pre/BIONET/preprocess/compare-neg/peaklist-neg-gz4_5-sort.txt'
if not os.path.exists(output_dir):
os.makedirs(output_dir)
class SpectrumProcessor:
def __init__(self, mz, intensity):
self.mz = mz
self.intensity = intensity
@staticmethod
def peak_picking(mz_array, intensity_array, snr=1, sigma=2.0):
"""
Identify peaks in the intensity array based on a signal-to-noise ratio (SNR) threshold.
:param mz_array: Array of m/z values.
:param intensity_array: Array of intensity values corresponding to the m/z values.
:param snr: Signal-to-noise ratio threshold for peak detection.
:param sigma: Standard deviation for Gaussian kernel used in smoothing.
:return: Arrays of m/z values and intensities for detected peaks.
"""
smoothed_intensity = gaussian_filter1d(intensity_array, sigma=sigma)
mean_intensity = np.mean(smoothed_intensity)
std_intensity = np.std(smoothed_intensity)
peaks = smoothed_intensity > (mean_intensity + snr * std_intensity)
return mz_array[peaks], intensity_array[peaks]
def binning(self, Da):
"""
Binning algorithm for spectra.
:return: 1D numpy array, binned spectrum.
"""
bin_size = int(Da / (self.mz[1] - self.mz[0]))
bin_spectrum = np.mean(
np.pad(self.intensity, (0, bin_size - len(self.intensity) % bin_size), mode='constant', constant_values=0)
.reshape(-1, bin_size),
axis=1)
mz_values1 = self.mz[::bin_size]
return mz_values1, bin_spectrum
parser = ImzMLParser(input_path)
coordinates = []
mz_values_list = []
intensity_values_list = []
for idx, (x, y, z) in enumerate(parser.coordinates):
mz_array, intensity_array = parser.getspectrum(idx)
mz_peaks, intensity_peaks = SpectrumProcessor.peak_picking(mz_array, intensity_array)
coordinates.append((x, y, z))
print(idx)
mz_values_list.append(mz_peaks)
intensity_values_list.append(intensity_peaks)
with ImzMLWriter(output_path) as writer:
for idx, (mz_peaks, intensity_peaks) in enumerate(zip(mz_values_list, intensity_values_list)):
x, y, z = coordinates[idx]
print(idx)
writer.addSpectrum(mz_peaks, intensity_peaks, (x, y, z))
def extract_imzml_peaks(imzml_file, output_dir):
parser = ImzMLParser(imzml_file)
for i, (x, y, z) in enumerate(parser.coordinates, start=1):
mzs, intensities = parser.getspectrum(i - 1)
output_filename = os.path.join(output_dir, f"peaks_{i:05d}.csv")
with open(output_filename, 'w', newline='') as csvfile:
csvwriter = csv.writer(csvfile)
csvwriter.writerow(['mz', 'intensity'])
for mz, intensity in zip(mzs, intensities):
csvwriter.writerow([mz, intensity])
extract_imzml_peaks(output_path, output_dir)
imzml = ImzMLParser(output_path)
coordinates = np.array(imzml.coordinates)[:, :2]
np.savetxt('/Users/ForRiver/OneDrive/Desktop/Pre/BIONET/preprocess/compare-neg/cood-gz4_5.csv', coordinates, delimiter=',')
def peak_filter(PATH, min_frequency=0.005, type='MATLAB', file=' '):
if type == 'MATLAB':
path = PATH
m = len(os.listdir(path))
mz = []
for i in range(1, m + 1):
data = pd.read_csv(path + str(i), header=None)
data = data.values
mz.extend(list(data[:, 0]))
mzs = sorted(set(np.around(mz, 4)))
print('all mz number is ', len(mzs))
dict_total = {}
for i in mzs:
dict_total[i] = 0
for i in range(m):
data = pd.read_csv(path, str(i + 1), header=None)
data = data.values
_u = np.around(data[:, 0], 4)
for j in _u:
dict_total[j] += 1
i = 0
peak_list = []
for key, value in dict_total.items():
if value > m * min_frequency:
i += 1
peak_list.append(key)
print('filter peak is ', len(peak_list))
np.savetxt('peak_list3', peak_list)
output_mat = np.zeros((m, len(peak_list)))
for i in range(m):
print('processing ', i, 'piex')
data = pd.read_csv(path, str(i + 1), header=None)
data = data.values
_u = np.around(data[:, 0], 4)
for j in range(len(_u)):
if _u[j] in peak_list:
output_mat[i, peak_list.index(_u[j])] = data[j, 1]
return output_mat
if type == 'R':
path = PATH
m = len(os.listdir(path))
mz = []
for i in range(1, m):
if i % 100 == 0:
print(i)
k = '{:0>5d}'.format(i)
data = pd.read_csv(path + file + k + '.csv', header=None)
data = data.values[1:]
mz.extend(list(data[:, 0]))
mz = [float(x) for x in mz]
mzs = list(set(mz))
print('all mz number is ', len(mzs))
dict_total = {}
for i in mzs:
dict_total[i] = 0
for i in range(1, m):
if i % 100 == 0:
print(i)
k = '{:0>5d}'.format(i)
data = pd.read_csv(path + file + k + '.csv', header=None)
data = data.values[1:]
dataint = [float(x) for x in data[:, 0]]
dataint2 = [float(x) for x in data[:, 1]]
_u = dataint
threhold = np.percentile(dataint2, 95) * 0.001
for j in range(len(_u)):
if dataint2[j] >= threhold:
dict_total[_u[j]] += 1
i = 0
peak_list = []
for key, value in dict_total.items():
if value > m * min_frequency:
i += 1
peak_list.append(key)
print('after filter peak is ', len(peak_list))
output_mat = np.zeros((m, len(peak_list)))
for i in range(1, m):
k = '{:0>5d}'.format(i)
if i % 100 == 0:
print(i)
data = pd.read_csv(path + file + k + '.csv', header=None)
data = data.values[1:]
dataint = [float(x) for x in data[:, 0]]
_u = dataint
for j in range(len(_u)):
if _u[j] in peak_list:
output_mat[i, peak_list.index(_u[j])] = data[j, 1]
return output_mat, peak_list
output_mat, peak_list = peak_filter(output_dir, min_frequency=0.05, type='R', file='peaks_')
spa_data = sparse.bsr_matrix(output_mat)
sparse.save_npz('/Users/ForRiver/OneDrive/Desktop/Pre/BIONET/preprocess/compare-neg/gz4_5.npz', spa_data)
np.savetxt('/Users/ForRiver/OneDrive/Desktop/Pre/BIONET/preprocess/compare-neg/peaklist-gz4_5', peak_list)
data0 = sparse.load_npz('/Users/ForRiver/OneDrive/Desktop/Pre/BIONET/preprocess/compare-neg/gz4_5.npz').toarray()
data = data0[1:, :]
coordinates = np.loadtxt('/Users/ForRiver/OneDrive/Desktop/Pre/BIONET/preprocess/compare-neg/cood-gz4_5.csv', delimiter=',')
x, y = coordinates[:, 0].astype(int), coordinates[:, 1].astype(int)
output_mat = np.zeros((np.max(y) + 1, np.max(x) + 1, data.shape[1]))
for i in range(len(data)):
output_mat[y[i], x[i]] = data[i]
output_mat = np.array([list(output_mat[i]) for i in range(len(output_mat)) if np.sum(output_mat[i]) != 0])
output_mat = np.array([list(output_mat[:, i]) for i in range(len(output_mat[0])) if np.sum(output_mat[:, i]) != 0])
data = sparse.bsr_matrix(output_mat.reshape(output_mat.shape[0] * output_mat.shape[1], output_mat.shape[2]))
sparse.save_npz('/Users/ForRiver/OneDrive/Desktop/Pre/BIONET/preprocess/compare-neg/gz4_5_%d_%d.npz' % (output_mat.shape[0], output_mat.shape[1]), data)
with open('/Users/ForRiver/OneDrive/Desktop/Pre/BIONET/preprocess/compare-neg/peaklist-gz4_5', 'r') as file:
lines = file.readlines()
lines = [line.strip() for line in lines]
lines.sort()
with open(sorted_peaklist_file, 'w') as file:
for line in lines:
file.write(f"{line}\n")
print("peaklist_saved")
data0 = sparse.load_npz(
'/Users/ForRiver/OneDrive/Desktop/Pre/BIONET/preprocess/compare-neg/gz4_5_%d_%d.npz' % (output_mat.shape[0], output_mat.shape[1])).toarray()
with open('/Users/ForRiver/OneDrive/Desktop/Pre/BIONET/preprocess/compare-neg/peaklist-gz4_5', 'r') as f:
reference = [float(x.strip()) for x in f.readlines()]
with open(sorted_peaklist_file, 'r') as f:
target = [float(x.strip()) for x in f.readlines()]
indices = [target.index(value) if value in target else -1 for value in reference]
data = np.vstack([data0[:, idx] for idx in indices if idx != -1]).T
np.save('/Users/ForRiver/OneDrive/Desktop/Pre/BIONET/preprocess/compare-neg/gz4_5', data)
print("Data has been saved.")
|